












网站维护
系统内容更新/升级中


In chapters 7 and 10[1], we discussed the use of LUMO and reaction energy profile for regioselectivity analyses of SnAr reactions of polyhalogenated aromatic compounds in general. In this chapter, we will discuss what we learned with polyhalogenated benzaldehyde substrates specifically.
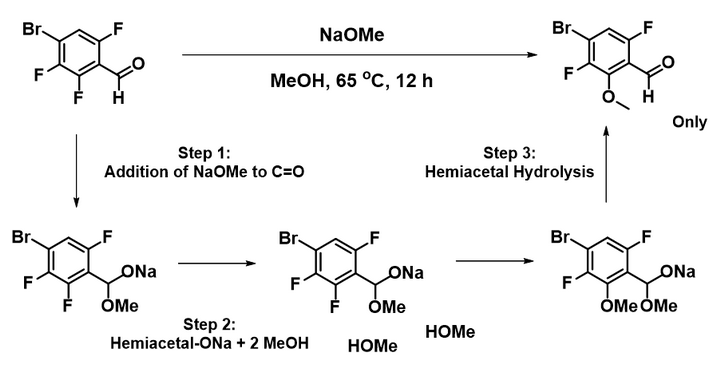
Shown in Figure 1 is the reaction of sodium methoxide with 2,3,6-trifluoro-4-bromo-benzaldehyde (1) with four potentially displaceable halogens. We learned from organic chemistry the following pattern F > Cl ≈ Br > I for leaving ability in SnAr reaction. We reasoned that the fluoro groups at C2 and C6 will be more labile than the bromo group on C4[2], and the aldehyde group enhances their reactivity. It is not obvious to us whether the reaction will be selective at C2 or C6, or provide a mixture of both products. Experimentally, we observed only C2 displacement. Why is the reaction so selective? Benzaldehyde 1 is quite electron deficient, why do we need to warm up the reaction to 65 ºC for it to proceed?
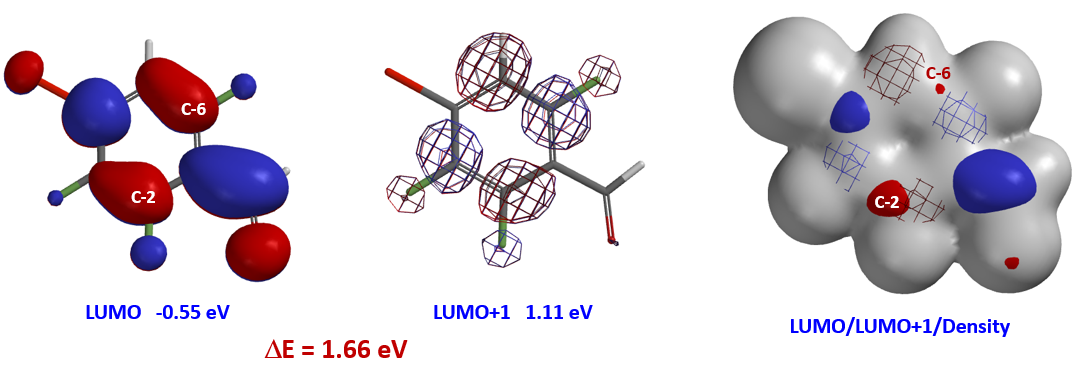
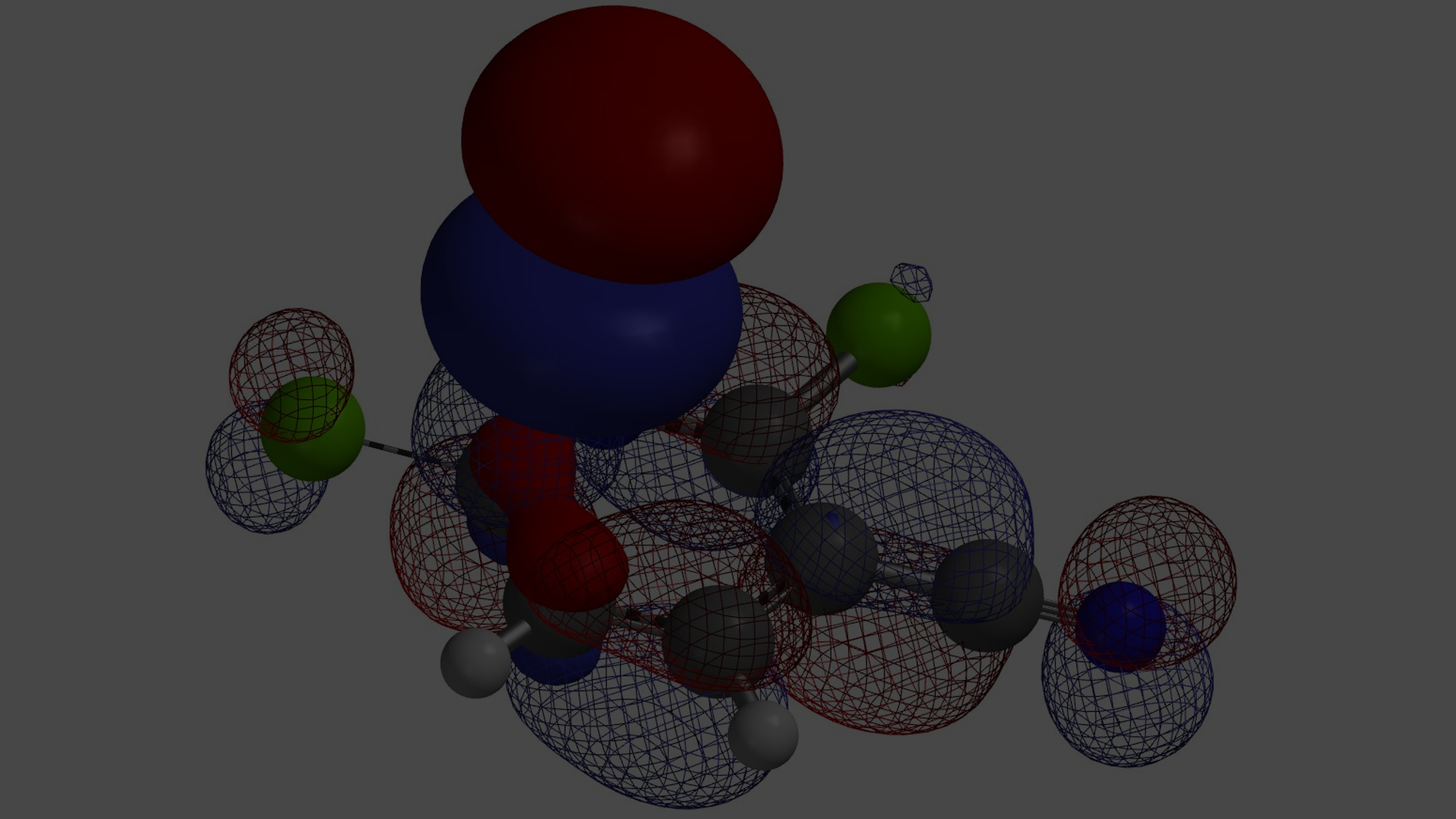
Shown in Figure 2 are LUMO (-0.55 eV) and LUMO+1 (1.11 eV) of benzaldehyde 1. Since the energy difference between them, ΔΕ = 1.66 eV, is relatively large (Figure 2), we could focus our analysis on LUMO. Its lobes on C2 and C6 are similar in size, yet after we overlaid Electron Density Map[3] on the orbital, it became obvious that the LUMO lobe at C2 is more accessible than the one at C6, accounting for the selectivity observed.
Next, we calculated for the reaction energy profile of the addition step at C2 and C6 positions toward formation of the corresponding Meisenheimer complexes.
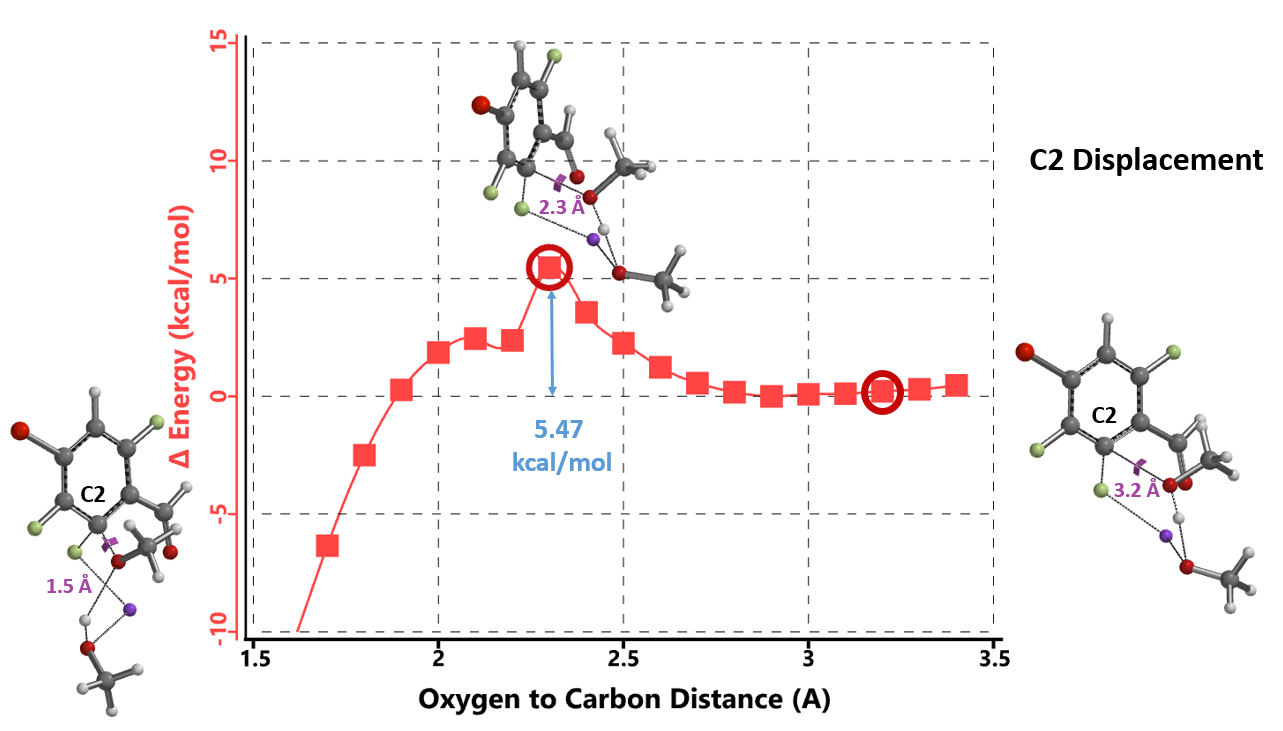
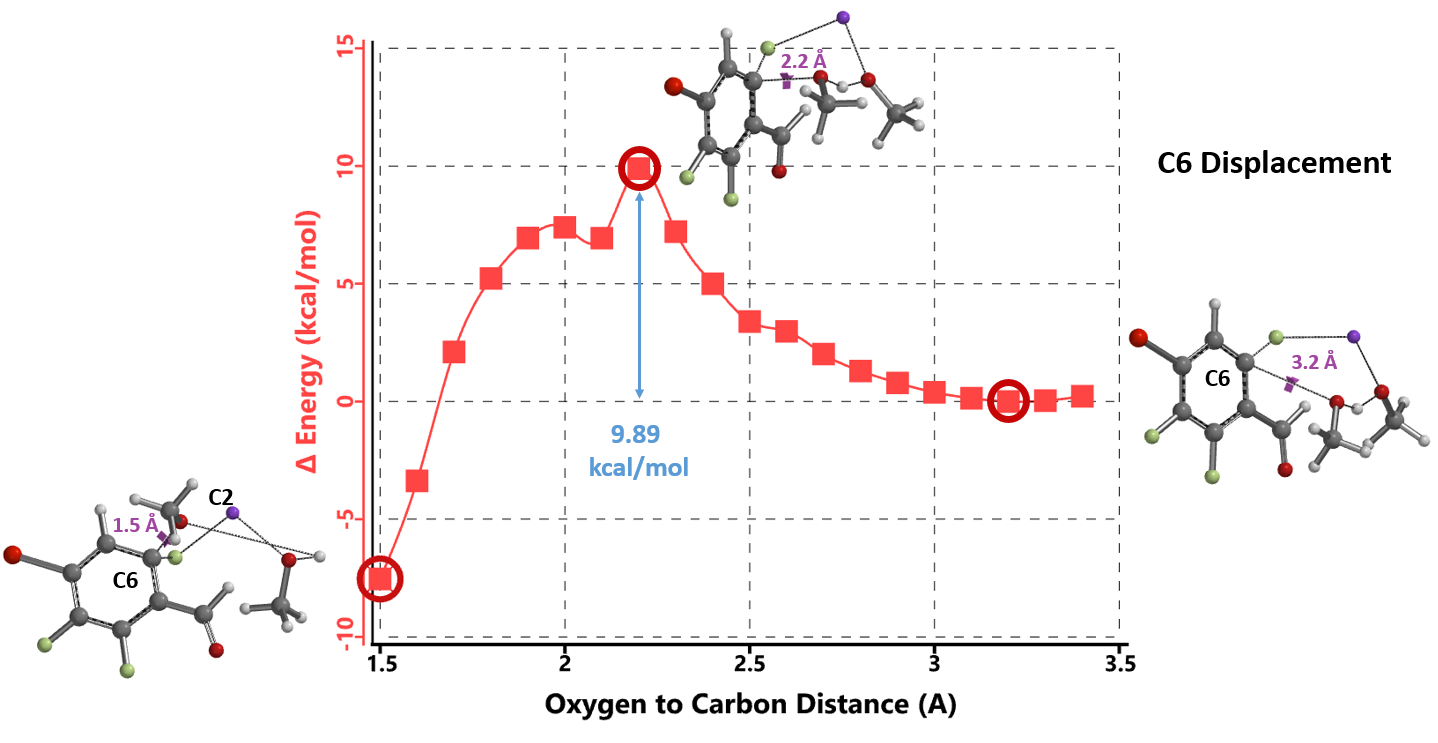
The results are shown in Figure 3. The activation energy required for addition at C6 position is 4.42 kcal/mol higher than that required for C2, suggesting that the reaction is more likely to occur at C2 position, consistent with the experimental results. The calculated activation energy of is relatively low at 5.47 kcal/mol, suggesting that the reaction could proceed at room temperature or lower, in contrast to the required experimental conditions (65 ºC, 12 hours).
Reviewing LUMO of substrate 1 (Figure 2), we found LUMO lobe on the aldehyde carbon is significantly larger than that on C2 and C6. Methoxide nucleophilic addition is more likely to occur preferentially at the aldehyde group first. Above assumed reaction mechanism of direct displacement could be an oversimplification.
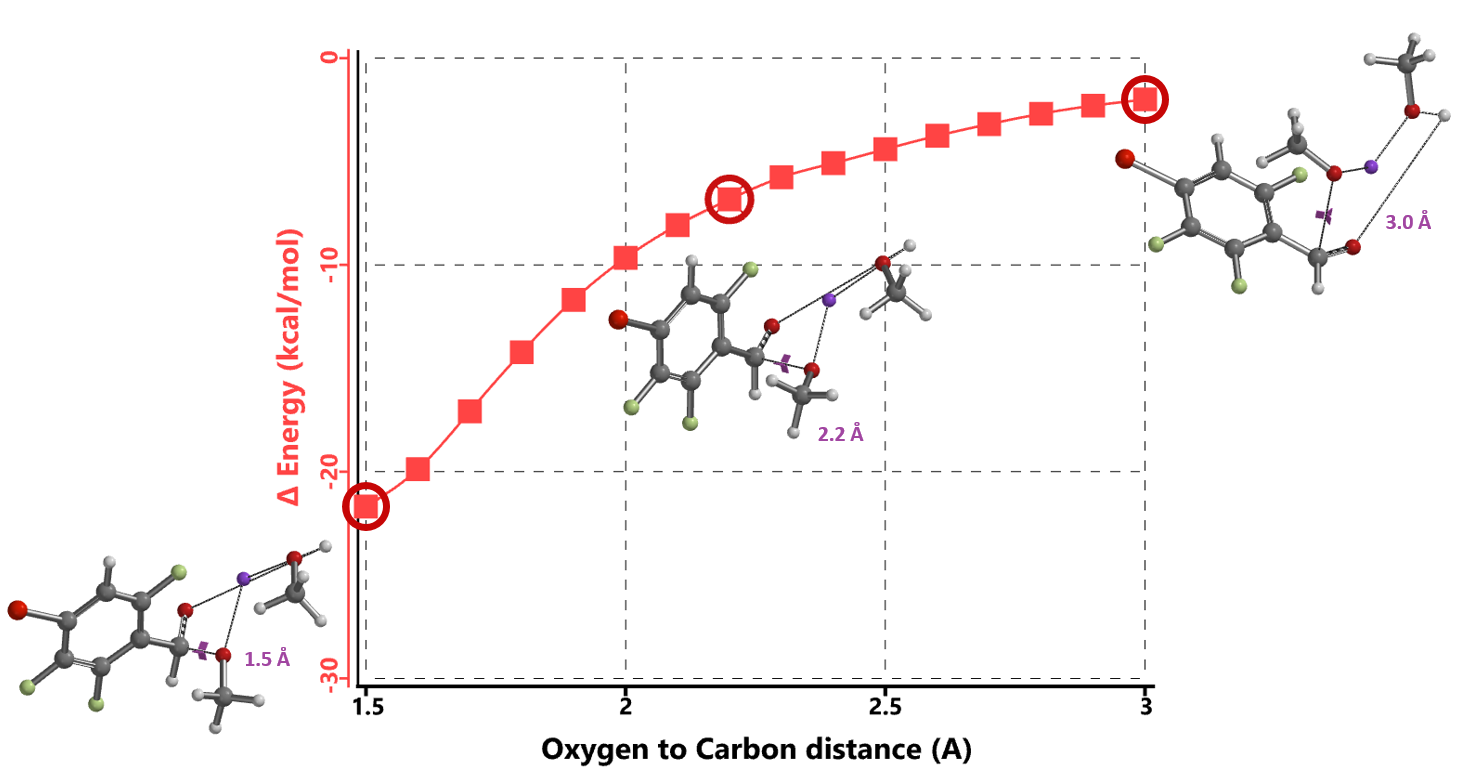
We reasoned that the addition of sodium methoxide to the aldehyde group precedes nucleophilic attack at the C-F carbons. Reaction energy profile calculated for the formation of the hemiacetal sodium salt (Figure 4) suggests that addition is exothermic and proceeds with no energy barrier, supporting the hypothesis that the hemiacetal sodium salt is the “actual” substrate for subsequent nucleophilic aromatic substitution.
Next, we calculated for reaction energy profile and transition state of the nucleophilic substitution. Based on structural characteristics of the hemiacetal salt, we evaluated two options, 1 MeOH and 2 MeOH models.
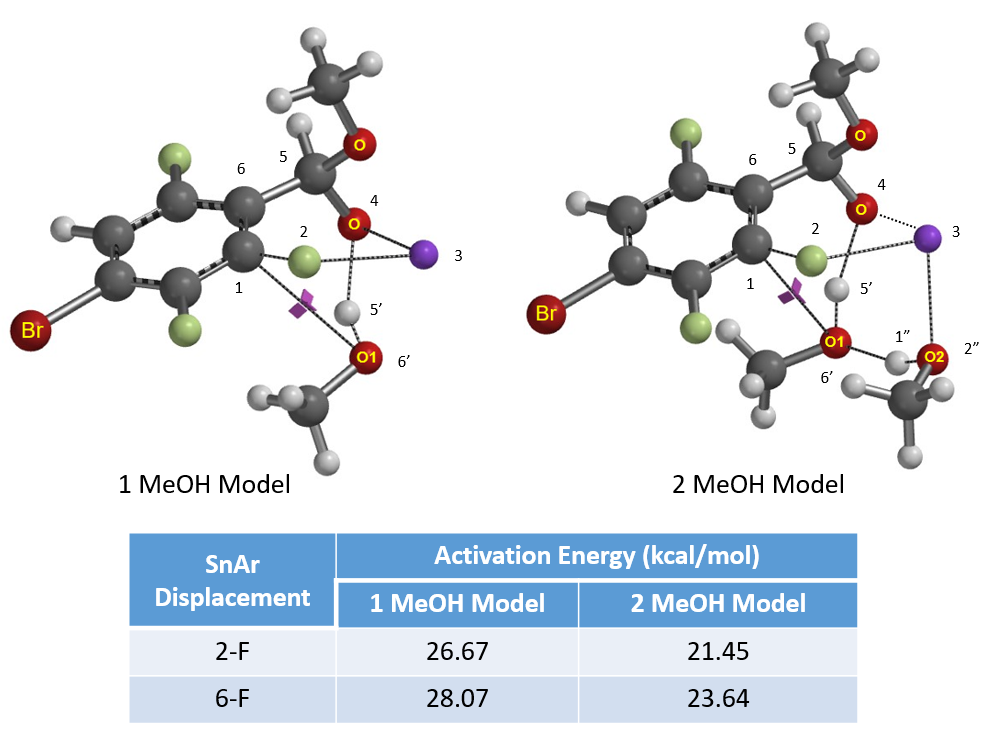
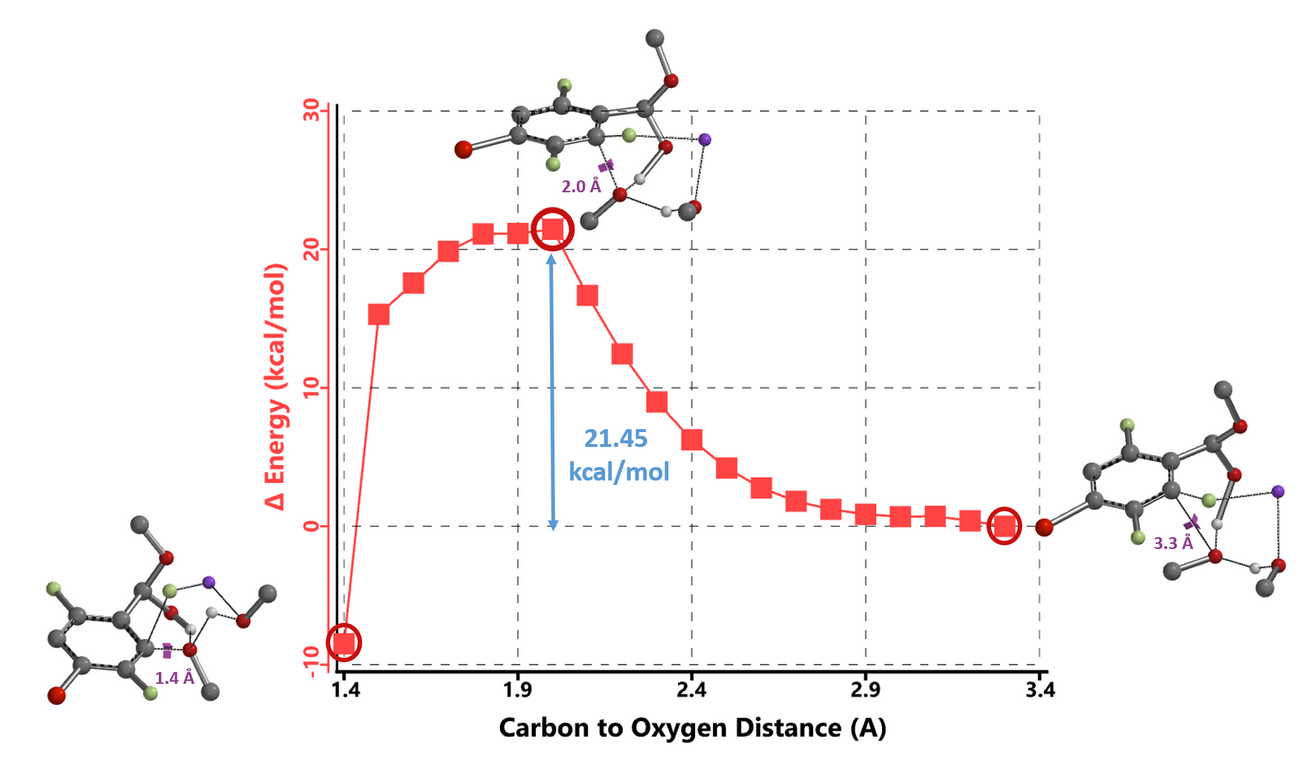
Activation energy for methoxide addition at C2 of benzaldehyde-methoxide adduct is 1.40 and 2.19 kcal/mol lower than attack at C6 for the 1 MeOH and 2 MeOH models, respectively (Figure 5), accounting for the selective C2 substitution. Reaction energy profile calculated with the 2 MeOH model exhibits unique gentle drop in energy between 2.0 to 1.5 Å (Figure 6, C2-F carbon to incoming methoxy oxygen)2c corresponding to an energy barrier of 21.45 kcal/mol, in better agreement with experimental conditions (65 ºC, 12 hours).
Transition state (iFreq i350 cm-1) and Intrinsic Reaction Coordinate calculated (not shown) are supportive of the above sodium hemiacetal salt 2 MeOH reaction model. We assume upon aqueous work up, the hemiacetal on the SNAr product hydrolyzes back to the aldehyde (Figure 7).
QM calculations suggest that SnAr reaction between 2,3,6-trifluoro-4-bromobenzaldehyde (1) and sodium methoxide proceeds via their corresponding hemiacetal product. Methanol solvent molecules are an integral part of the transition state. Both 1 MeOH and 2 MeOH reaction models could account for preferential formation of C2 displacement product 2b. Calculated activation energy of 21.45 kcal/mol with the 2 MeOH model is in better agreement with experimental conditions.
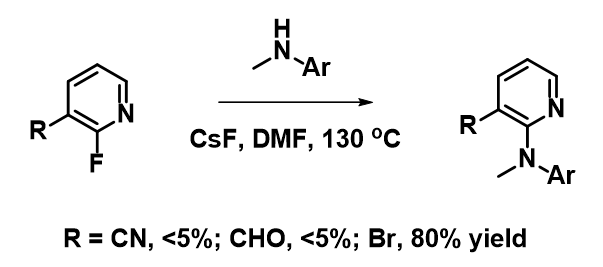
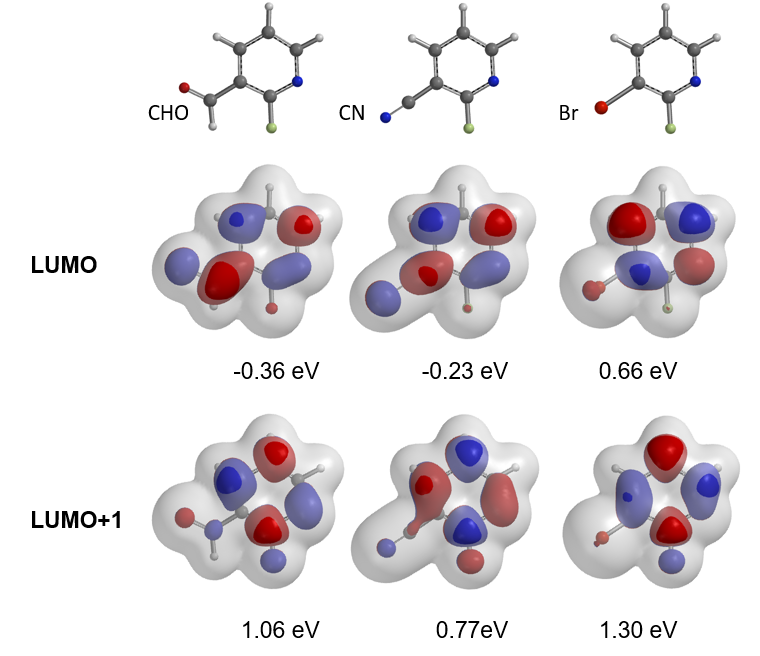
For the SNAr reaction of 2-fluoropyridine analogs, electron withdrawing groups, such as CN and CHO, at C3 position, were anticipated to increase the electrophilicity of the substrates and accelerate the SNAr reaction (Figure 8). However, less than 5% conversions were observed, and most of the starting materials were recovered. On the other hand, SNAr of the bromo analog proceeded smoothly with an isolated yield of 80%. How could we account for these? Below are overlays of LUMO and LUMO+1 of the pyridine analogs with their electron density maps.
This article is written and edited by Dajin Tan, Yongsheng Chen, Liting Dong, John S. Wai
References:
[1] (a) QM Chapter 7: LUMO Analysis of Electrophiles (Part II). (b) QM Chapter 10: SNAr Reaction of Polyhalogenated Heterocycles
[2] (a) S. Rohrbach, A.J. Smith, J.H. Pang, D.L. Poole, T. Tuttle, S. Chiba S, J.A. Murphy, Angew. Chem. Int. Ed., 2019, 58, 16368. (b) O. Acevedo, W.L. Jorgensen, Org. Lett., 2004, 6, 2881. (c) E.E. Kwan, Y. Zeng, H.A. Besser, E.N. Jacobsen, Nature Chemistry, 2018, 10, 917.
[3] Spartan’20 Tutorial and User’s Guide (2020). Irvine, CA, USA: Wavefunction, Inc. pp362-368.